









Contidos
Esclerose tuberosa de Bourneville
Que é?
A esclerose tuberosa de Bourneville é unha enfermidade xenética complexa caracterizada polo desenvolvemento dun tumor benigno (non canceroso) en diferentes partes do corpo. Estes tumores poden localizarse na pel, o cerebro, os riles e outros órganos e tecidos. Esta patoloxía tamén pode causar serios problemas no desenvolvemento do individuo. Non obstante, as manifestacións clínicas e a gravidade da enfermidade varían dun paciente a outro.
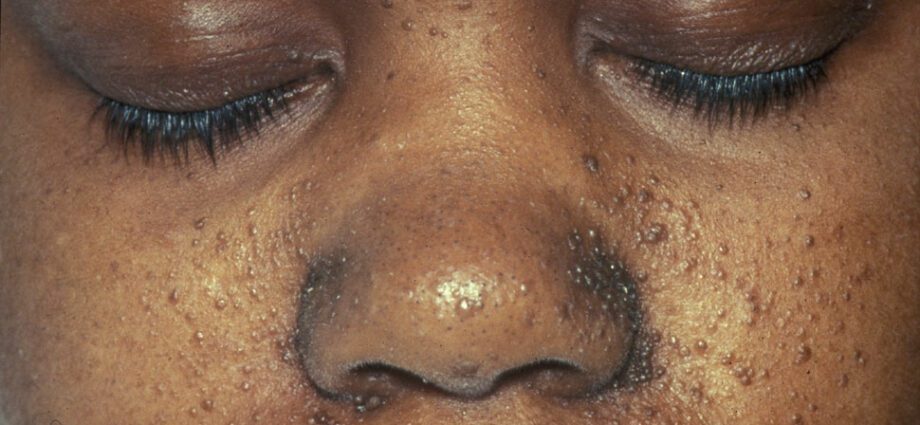
As anomalías da pel asociadas son xeralmente similares ás manchas da pel ou ás zonas onde a pel é máis clara que no resto do corpo. O desenvolvemento de tumores na cara chámase angiofibroma.
No contexto do dano cerebral, os signos clínicos son crises epilépticas, problemas de comportamento (hiperactividade, agresividade, discapacidade intelectual, problemas de aprendizaxe, etc.). Algúns nenos con a enfermidade incluso teñen algún tipo de autismo, trastornos do desenvolvemento, que afectan as interaccións sociais e a comunicación. Os tumores cerebrais benignos tamén poden causar complicacións que poden ser mortais para o suxeito.
O desenvolvemento de tumores nos riles é común en persoas con esclerose tuberosa. Isto pode causar complicacións graves na función renal. Ademais, os tumores poden desenvolverse no corazón, pulmóns e retina. (2)
É unha enfermidade rara, cuxa prevalencia (número de casos nunha determinada poboación nun momento determinado) ascende a 1/8 a 000/1 persoas. (15)
os síntomas
As manifestacións clínicas asociadas á esclerose tuberosa de Bourneville varían segundo os órganos afectados. Ademais, os síntomas asociados á enfermidade varían moito dun individuo a outro. Con síntomas que van de leves a graves.
Entre os síntomas máis identificados desta enfermidade destacan as crises epilépticas, os trastornos cognitivos e do comportamento, as alteracións cutáneas, etc. Os órganos máis afectados son: o cerebro, o corazón, os riles, os pulmóns e a pel.
O desenvolvemento de tumores malignos (cancerosos) é posible nesta enfermidade, pero son raros e afectan principalmente aos riles.
Os signos clínicos da enfermidade no cerebro orixínanse de ataques a diferentes niveis:
- danos aos tubérculos corticais;
– nódulos ependimarios (SEN);
– astrocitomas ependimarios xigantes.
Ten como consecuencia: o desenvolvemento do retraso mental, dificultades de aprendizaxe, trastornos da conduta, agresividade, trastornos de atención, hiperactividade, trastornos obsesivo-compulsivos, etc.
O dano renal caracterízase polo desenvolvemento de quistes ou angiomiolipomas. Estes poden provocar dor renal e incluso insuficiencia renal. Se se nota un sangrado abundante, pode ser por anemia grave ou presión arterial alta. Tamén poden ser visibles outras consecuencias máis graves pero raras, en particular o desenvolvemento de carcinomas (tumor das células constituíntes do epitelio).
O dano ocular pode ser semellante aos puntos visibles da retina, causando trastornos visuais ou mesmo cegueira.
As anomalías da pel son numerosas:
– máculas hipomelánicas: que dan lugar á aparición de manchas claras na pel, en calquera parte do corpo, como consecuencia dunha deficiencia de melanina, proteína que dá cor á pel;
- aparición de manchas vermellas na cara;
- manchas descoloridas na fronte;
– outras anomalías da pel, dependentes dun individuo a outro.
As lesións pulmonares están presentes en 1/3 dos pacientes cun lixeiro predominio feminino. Os síntomas asociados son entón dificultades respiratorias máis ou menos graves.
As orixes da enfermidade
A orixe da enfermidade é xenética e hereditaria.
A transmisión implica mutacións nos xenes TSC1 e TSC2. Estes xenes de interese entran en xogo na formación de proteínas: hamartina e tuberina. Estas dúas proteínas permiten, mediante un xogo interactivo, regular a proliferación celular.
Os pacientes con enfermidade nacen con polo menos unha copia mutada destes xenes en cada unha das súas células. Estas mutacións limitan entón a formación de hamartina ou tubertina.
No contexto onde se mutan as dúas copias do xene, impiden completamente a produción destas dúas proteínas. Esta deficiencia proteica xa non permite que o organismo regule o crecemento de determinadas células e, neste sentido, leva ao desenvolvemento de células tumorais en diferentes tecidos e/ou órganos.
Os factores de risco
Os factores de risco para desenvolver tal patoloxía son xenéticos.
De feito, a transmisión da enfermidade é efectiva a través dun modo autosómico dominante. Ou ben, o xene mutado de interese sitúase nun cromosoma non sexual. Ademais, a presenza de só unha das dúas copias do xene mutado é suficiente para que se desenvolva a enfermidade.
Neste sentido, un individuo que posúe un destes dous pais que padece a enfermidade ten un 50% de risco de desenvolver o propio fenotipo enfermo.
Prevención e tratamento
O diagnóstico da enfermidade é en primeiro lugar diferencial. Baséase en criterios físicos atípicos. Na maioría dos casos, os primeiros signos característicos da enfermidade son: a presenza de ataques epilépticos recorrentes e atrasos no desenvolvemento do suxeito. Noutros casos, estes primeiros signos dan lugar a manchas na pel ou a identificación dun tumor cardíaco.
Tras este primeiro diagnóstico, son imprescindibles exames adicionais para validar ou non o diagnóstico. Estes inclúen:
- unha exploración cerebral;
– unha resonancia magnética (MRI) do cerebro;
– unha ecografía do corazón, do fígado e dos riles.
O diagnóstico pode ser eficaz no nacemento do neno. En caso contrario, é importante que se realice o máis rápido posible para facerse cargo do paciente canto antes.
Actualmente, non hai cura para a enfermidade. Polo tanto, os tratamentos asociados son independentes dos síntomas que presenta cada individuo.
Normalmente, se administran medicamentos antiepilépticos para limitar as convulsións. Ademais, tamén se prescriben medicamentos para o tratamento de células tumorais do cerebro e dos riles. No contexto dos problemas de comportamento, é necesario un tratamento específico do neno.
O tratamento da enfermidade adoita ser a longo prazo. (1)