









Contidos
Enfermidade de Wilson
Que é?
A enfermidade de Wilson é unha enfermidade xenética hereditaria que impide a eliminación do cobre do corpo. A acumulación de cobre no fígado e no cerebro causa problemas hepáticos ou neurolóxicos. A prevalencia da enfermidade de Wilson é moi baixa, ao redor de 1 de cada 30 persoas. (000) Existe un tratamento eficaz para esta enfermidade, pero o seu diagnóstico precoz é problemático porque permanece en silencio durante moito tempo.
os síntomas
A acumulación de cobre comeza no nacemento, pero os primeiros síntomas da enfermidade de Wilson adoitan non aparecer ata a adolescencia ou a idade adulta. Poden ser moi diversos porque varios órganos están afectados pola acumulación de cobre: corazón, riles, ollos, sangue... Os primeiros signos son hepáticos ou neurolóxicos en tres cuartas partes dos casos (40% e 35% respectivamente), pero poden tamén ser psiquiátrico, renal, hematolóxico e endocrinolóxico. O fígado e o cerebro están especialmente afectados porque xa conteñen de forma natural a maior parte de cobre. (2)
- Trastornos hepáticos: ictericia, cirrose, insuficiencia hepática...
- Trastornos neurolóxicos: depresión, trastornos do comportamento, dificultades de aprendizaxe, dificultades para expresarse, tremores, calambres e contracturas (distonía)...
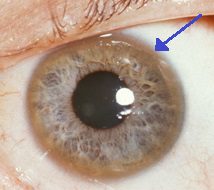
O anel Keyser-Fleisher que rodea o iris é característico da acumulación de cobre no ollo. Ademais destes síntomas agudos, a enfermidade de Wilson pode presentar síntomas pouco característicos, como fatiga xeral, dor abdominal, vómitos e perda de peso, anemia e dor nas articulacións.
As orixes da enfermidade
Na orixe da enfermidade de Wilson, hai unha mutación no xene ATP7B situado no cromosoma 13, que está implicado no metabolismo do cobre. Controla a produción dunha proteína ATPase 2 que xoga un papel no transporte de cobre do fígado a outras partes do corpo. O cobre é un bloque de construción necesario para moitas funcións celulares, pero en exceso de cobre vólvese tóxico e dana os tecidos e órganos.
Os factores de risco
A transmisión da enfermidade de Wilson é autosómica recesiva. Polo tanto, é necesario recibir dúas copias do xene mutado (do pai e da nai) para desenvolver a enfermidade. Isto significa que homes e mulleres están igualmente expostos e que dous pais portadores do xene mutado pero non enfermos teñen o risco de transmitir a enfermidade de cada catro en cada nacemento.
Prevención e tratamento
Existe un tratamento eficaz para deter a progresión da enfermidade e reducir ou mesmo eliminar os seus síntomas. Tamén é necesario que se inicie precozmente, pero moitas veces son necesarios moitos meses despois da aparición dos síntomas para diagnosticar esta enfermidade silenciosa, pouco coñecida e cuxos síntomas apuntan a moitas outras afeccións (hepatite para a que é dano hepático e depresión por afectación psiquiátrica) .
Un tratamento "quelante" permite atraer o cobre e eliminalo na urina, limitando así a súa acumulación nos órganos. Está baseado na D-penicilamina ou Trientine, medicamentos que se toman por vía oral. Son eficaces, pero poden provocar efectos secundarios graves (danos nos riles, reaccións alérxicas, etc.). Cando estes efectos secundarios son demasiado importantes, recorremos á administración de zinc que limitará a absorción de cobre polos intestinos.
Un transplante de fígado pode ser necesario cando o fígado está demasiado danado, que é o caso do 5% das persoas con enfermidade de Wilson (1).
Ofrécese unha proba de detección xenética aos irmáns dunha persoa afectada. Dá lugar a un tratamento preventivo eficaz no caso de que se detecte unha anomalía xenética no xene ATP7B.