









Contidos
Enfermidade de Huntington
Que é?
A enfermidade de Huntington é unha enfermidade neurodexenerativa xenética e herdada. Ao destruír as neuronas en certas áreas do cerebro, provoca trastornos motores e psiquiátricos graves e pode provocar unha completa perda de autonomía e morte. O xene cuxa alteración causa a enfermidade identificouse nos anos 90, pero a enfermidade de Huntington segue sendo incurable ata os nosos días. Afecta a unha de cada 10 persoas en Francia, o que representa ao redor de 000 pacientes.
os síntomas
Ás veces chámaselle "corea de Huntington" porque o síntoma máis característico da enfermidade son os movementos involuntarios (chamados coreicos) que causa. Non obstante, algúns pacientes non presentan trastornos coreicos e os síntomas da enfermidade son máis amplos: a estes trastornos psicomotores engádense frecuentemente trastornos psiquiátricos e de conduta. Estes trastornos psiquiátricos que se producen con frecuencia ao comezo da enfermidade (e ás veces aparecen antes dos trastornos motores) poden provocar demencia e suicidio. Os síntomas normalmente aparecen ao redor dos 40-50 anos, pero obsérvanse formas temperás e tardías da enfermidade. Nótese que todos os portadores do xene mutado un día declaran a enfermidade.
As orixes da enfermidade
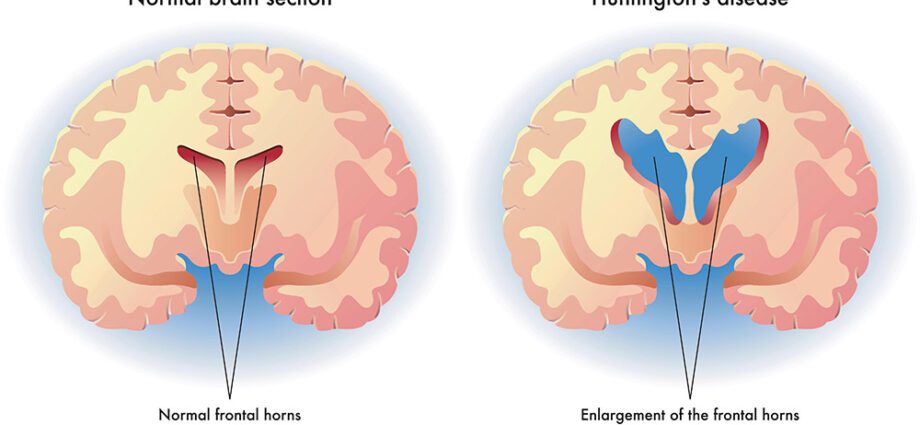
O médico estadounidense George Huntington describiu a enfermidade de Huntington en 1872, pero non foi ata 1993 cando se identificou o xene responsable. Localizouse no brazo curto do cromosoma 4 e nomeouse IT15. A enfermidade é causada por unha mutación neste xene que controla a produción da proteína huntingtina. A función precisa desta proteína aínda é descoñecida, pero sabemos que a mutación xenética faina tóxica: provoca depósitos no medio do cerebro, máis precisamente no núcleo de neuronas do núcleo caudado, logo da cortiza cerebral. Non obstante, hai que ter en conta que a enfermidade de Huntington non está sistematicamente ligada á IT15 e pode ser causada pola mutación doutros xenes. (1)
Os factores de risco
A enfermidade de Huntington pódese transmitir de xeración en xeración (chámase "autosómica dominante") e o risco de transmisión á descendencia é de un en dous.
Prevención e tratamento
O cribado xenético da enfermidade en persoas con risco (con antecedentes familiares) é posible, pero moi supervisado pola profesión médica, porque o resultado da proba non está exento de consecuencias psicolóxicas.
O diagnóstico prenatal tamén é posible, pero está estritamente enmarcado pola lei, porque suscita cuestións de bioética. Non obstante, unha nai que estea a considerar a interrupción voluntaria do embarazo no caso de que o seu feto leve o xene alterado ten dereito a solicitar este diagnóstico prenatal.
Ata a data, non existe tratamento curativo e só o tratamento dos síntomas pode aliviar a persoa enferma e retardar o seu deterioro físico e psicolóxico: psicofármacos para aliviar os trastornos psiquiátricos e os episodios de depresión que a miúdo van parellos á enfermidade. ; medicamentos neurolépticos para reducir os movementos coreicos; rehabilitación mediante fisioterapia e logopedia.
A busca de futuras terapias diríxese cara ao transplante de neuronas fetais co fin de estabilizar as funcións motoras do cerebro. En 2008, investigadores do Instituto Pasteur e do CNRS demostraron a capacidade do cerebro para auto-repararse identificando unha nova fonte de produción de neuronas. Este descubrimento suscita novas esperanzas para o tratamento da enfermidade de Huntington e doutras enfermidades neurodexenerativas, como a enfermidade de Parkinson. (2)
Os ensaios de terapia xénica tamén están en marcha en varios países e están movéndose en varias direccións, unha delas é bloquear a expresión do xene hunttin mutado.